转自贾壮老师博客:http://blog.sciencenet.cn/blog-794272-718384.html
待整理
CHARMM力场提供了很多生物分子的力场参数,如蛋白质、脂质、核酸等,Gromacs中自带了CHARMM27力场,当然也可在Gromacs官网上下载到已转换成Gromacs支持的CHARMM36力场。不过使用Gromacs做MD模拟的朋友如果想模拟一个Gromacs中没有定义力场参数的分子,如糖,就需要手动为Gromacs添加相关力场参数了。这些力场参数多数从文献中得到,并且一般都是CHARMM格式的,即.prm 和 .rtf 格式,如何将它们转换成Gromacs支持的格式呢?这篇文章会详细讲解转换过程,并且给出python脚本以供参考。
首先分别讲解一下CHARMM软件和Gromacs软件如何完整定义一个分子的力场。在CHARMM软件中,主要通过2个文件来实现:.rtf 和.prm。
.rtf 文件定义了原子类型、各类型原子的质量、原子偏电荷、分子的键连接以及氢键等,它的格式如图1。MASS是用来定义原子类型的,每一行都定义了一种原子类型。
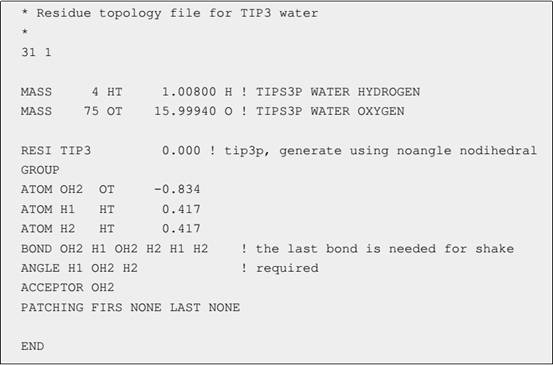
图__1. TIP3__水分子的_ .rtf 文件,CHARMM__中“!__”_后面的内容是注释
MASS后面跟着原子编号、原子类型名、原子质量和元素符号。GROUP语句用来定义一个残基(也可以是单独的分子):ATOM定义了残基中各原子的名字、所属的原子类型和该原子所带的偏电荷;BOND定义残基中原子的键连接情况,以原子对形式列出,如图1中的水分子定义了3个键OH2-H1、OH2-H2和H1-H2(最后一个键是SHAKE限制算法用到的);ANGLE定义键角;DONOR和ACCEPTOR分别用来定义氢键供体和受体。
.prm文件中定义了与能量相关的参数,如键伸缩能,键角、二面角的力常数以及范德华参数,图2给出了TIP3水分子的 .prm 文件。
BONDS数据块是用来定义键伸缩振动能量的,用简谐振动来近似:

BONDS数据块每行有4个字段,前两个是成键原子的类型,后两个分别是公式中的Kb(kcal/Å2/mol)和b0(Å)。
ANGLES数据块用来定义键角振动,同样是用简谐振动来近似:

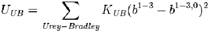
ANGLES数据每行有5或7个字段,后者是加了Urey-Bradley项的。前3项是形成键角的原子的类型,后面4项分别对应公式中的Kθ(kcal/度2/mol)、θ0(度)、KUB(kcal/Å2/mol)和b1-3,0(Å)。
关于Urey-Bradley项的解释可以查看gromacs手册:http://jerkwin.github.io/GMX/GMXman-4/#428-urey-bradley势
DIHEDRALS数据块用来定义二面角振动,用余弦函数来近似:
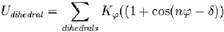
DIHEDRALS数据块每行有7个字段,前4项是形成二面角的原子的类型,后面3项分别对应公式中的Kφ(kcal/mol)、n(无单位)和δ(度)。

图__2. TIP3__水分子的_ .prm _文件
IMPROPER数据块用来保证分子中原子共平面的,用简谐振动近似:
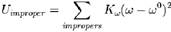
IMPROPER数据块每行有7个字段,前4项是形成二面角的原子的类型,第5项对应公式中的Kω(kcal/度2/mol),第6项一般是0,暂时不知道干嘛用的,第7项对应公式中的ω0(度)。
NONBONDED数据块用来定义非键作用,其中静电力可通过原子偏电荷求得,不需要额外参数,范德华力通常用L-J形式来描述:

通常力场中只定义单个原子的ε和rmin,成对原子的范德华力参数可以通过组合规则(combination rule)获得,常用的组合规则是εi,j = sqrt(εi×εj),rmini,j = (rmini+ rminj)/2。另外CHARMM力场将1-4作用单独分出来描述,也采用L-J形式,只是ε和rmin值有所不同。NONBONDED数据块每行有7个字段组成,第1个字段是原子类型,第2、5字段标识为ignored,通常是0,暂不知道干嘛用的,第3,4字段分别为vdw力的ε(kcal/mol)和rmin/2(Å),第6,7字段分别为1-4作用的ε(kcal/mol)和rmin/2(Å)。这里可以提前说明一下,Gromacs中1-4作用是以原子对的形式定义的,叫做pairtypes,转换时需要将CHARMM中的1-4作用通过组合规则两两组合生成pairtypes,这在后面还会详细讲到。
下面讲一下Gromacs中力场文件的格式。Gromacs主要通过5个文件来定义:.rtp、.hdb、atomtypes.atp、ffbonded.itp和ffnonbonded.itp。
.rtp文件用来定义残基(或分子)中原子所属的类型、原子偏电荷、键连接以及共平面原子,其余如键角和二面角是在这里不是必须的,程序一般通过原子类型生成,它将出现在ffbonded.itp文件中。图3是 .rtp 文件的一个示例,里面的注释很详尽,唯一需要说

_图__3. 甘氨酸的 .rtp 文件,Gromacs__中“;__”_后的内容是注释
明的是chargegroup项,同一个chargegroup中的原子偏电荷变化是以相同比例进行的,对于一个新加入的分子可简单的将每个原子划分到不同的chargegroup中。
.hdb文件是用来给残基(或分子)加氢的,我们知道用X-衍射得到的蛋白结构中一般没有氢,pdb2gmx命令会查询.hdb文件中定义的加氢规则为蛋白加氢。如果你的小分子中氢原子已在 .rtp文件中定义了,就不需要用 .hdb文件来加氢。因为一般力场中都会详细定义蛋白各种参数,我们只需添加一些小分子,而小分子中的氢原子多在 .rtp文件中定义,很少用到 .hdb文件,所以这里不再讲 .hdb文件格式。
atomtypes.atp文件中定义了力场中用到的所有原子类型,格式很简单,第一列是原子类型名,第二列是原子质量,如果你添加的分子用到了新的原子类型,就需要在这里添加相关信息。
ffbonded.itp文件定义了力场中的键作用:键伸缩振动、键角、二面角振动(func=9)

图__4. ffbonded.itp__文件格式
以及IMPROPER(在Gromacs中称为dihedraltypes(func=2))。它们定义及算法与CHARMM中相同,只是公式的形式以及单位有差异:
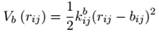
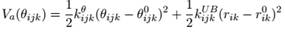


公式的差异表现在键长、键角、IMPROPER的简谐振动公式在Gromcas中多了一个系数1/2;单位的差异表现在CHARMM中能量单位为kcal,而Gromcas中使用kJ,CHARMM中长度单位为Å,而Gromacs中使用nm。参数从CHARMM转换到Gromacs规则如下:
(1) 键伸缩振动:Kgromacs= 2×Kcharmm×cal2j×100 (即Kgromacs = 836.8×Kcharmm,其中cal2j =4.184是cal转换到J的转换系数);b0gromacs = b0charmm/10。
(2) 键角振动:Kθgromacs = 2×Kθcharmm×cal2j(即Kθgromacs = 8.368×Kθcharmm);θ0不变;KUBgromacs = 2×KUBcharmm×cal2j×100(即KUBgromacs = 836.8×KUBcharmm);bUgromacs = bUcharmm/10。
(3) 二面角振动:Kφgromacs = Kφcharmm×cal2j(即Kφgromacs = 4.184×Kφcharmm);n和φ0不变。
(4) IMPROPER:Kξgromacs = 2×Kξcharmm×cal2j(即Kξgromacs = 8.368×Kξcharmm);ξ0不变。
值得注意的是这些参数在文件中位值不同,一定要弄清哪一列放哪个参数!
ffnonbonded.itp定义了力场中的非键作用,因静电力可通过原子偏电荷和coulomb公式求的,所以这个文件主要定义范德华力(在atomtypes数据块中)。与CHARMM类似,Gromacs也对1-4作用进行单独处理,处理方式与CHARMM完全相同,1-4作用定义在pairtypes数据块中。Gromacs中描述范德华力的L-J公式与CHARMM中也有差异:
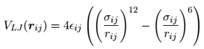
在转换之前我们需将CHARMM和Gromacs中的L-J公式化成相同的形式:

非键作用的参数转换规则如下:
εgromacs = -εcharmm×cal2j(即εgromacs = -4.184×εcharmm,不知道问什么CHARMM中加了负号)
σgromacs = 2×σcharmm/10/21/6(即σgromacs = 0.1781797436×σcharmm,CHARMM中记录的是rmin/2)
对于1-4作用,转换规则同上,只是Gromacs中通过组合规则(上文讲CHARMM时有提到)以原子对的形式记录在pairtypes数据块中,这需要自己计算添加。图5给出了一个转换示例,注意pairtypes是通过两两组合计算得到的,不区分先后,共6对。
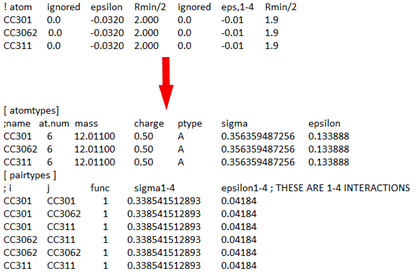
_图__5. _非键作用转换示例
对于简单分子可以手动转换并添加到Gromacs力场中,但是当分子较为复杂时手动转换将会变得非常繁琐并且容易出错,这时候就需要用脚本来进行批量处理。附件提供了用python写的转换脚本:
cvt_bd.py: 键伸缩项转换,输入文件bonds,输出文件bonds.out
cvt_agl.py: 键角转换,输入文件angles,输出文件angles.out
cvt_dihedral.py: 二面角转换,输入文件dihedrals,输出文件dihedrals.out
cvt_improper.py: improper项转换,输入文件impropers,输出文件impropers.out
cvt_nb.py: 非键作用转换,输入文件nobonded,输出文件vdw.out, pair.out
使用方法:
将.prm文件按数据块分成bonds(记录BONDS数据块)、angles(记录ANGLES数据块)、dihedrals(记录DIHEDRALS数据块)、impropers(记录IMPROPER数据块)和nonbonded(记录NONBONDED数据块)5个文件,然后按下面步骤处理:
(1)删除关键字BONDS、ANGLES、DIHEDRALS、IMPROPER和NONBONDED所在的行。
(2)删除“!”及其后面的注释内容,这可以在vim中使用命令:
:%s/!.*//
(3)删除行末多余的空格,可用vim命令:
:%s/ *$//
(4)删除空行,vim命令:
:g/^$/d
处理后的文件应该是每行具有相同列数(angles和nonbonded各行列数不全相同),格式一致的文本。然后运行上面的python脚本,将输出文件拷贝到ffbonded.itp和ffnonbonded.itp文件中对应的数据块内:
(1)bonds.out内容拷贝到ffbonded.itp文件[bondtypes]数据域中
(2)angles.out内容拷贝到ffbonded.itp文件[angletypes]数据域中
(3)dihedrals.out内容拷贝到ffbonded.itp文件[dihedraltypes] (func=9)数据域中
(4)impropers.out内容拷贝到ffbonded.itp文件[dihedraltypes](func=2)数据域中
(5)vdw.out内容拷贝到ffnonbonded.itp文件[atomtypes]数据域中
(6)pair.out内容拷贝到ffnonbonded.itp文件[pairtypes]数据域中
(7)vdw.out的1、3列内容拷贝到atomtypes.atp文件中
最后根据 .rtf 文件中记录的残基(或分子)原子类型、偏电荷、键连接和IMPROPER信息,按照Gromacs的 .rtp文件格式制做该残基(或分子)的 .rtp文件,将该文件放到相应的力场文件目录下(即ffbonded.itp所在目录)。最后提醒大家,分子的pdb文件一定要与 .rtp文件原子顺序和命名完全一致!
以上所有操作都正确的话就可以用Gromacs模拟该分子了,Happy simulating!