此文档不完全翻译自官方从头预测文档 当蛋白与已知解析蛋白结构为低相似性的时候从头预测变得非常有用。在从头预测算法中,蛋白序列在 n-mer 片段文库中进行phi/psi角采样来进行构象的扩展(extended conformation)和“折叠”。当整个蛋白质氨基酸残基数小于100以内时,其结果较为精确。
官方有Rosetta@home计划的在线服务器,可以直接提交序列,然后在线服务器进行计算,但每个帐号只能计算一条序列,且要注意的是不能进行已知序列或者高相似度序列的计算,否则会ban ip,删号。因为从头结构预测时间太久太长。 官方地址如下:http://robetta.bakerlab.org/ 网站截图如下:
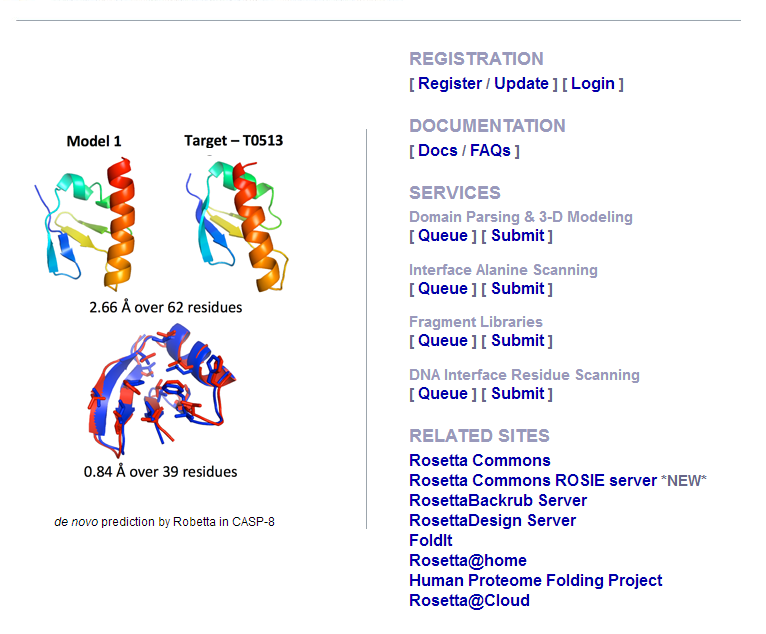
网站截图
该项目是伯克利分校搞的一个分布式系统项目BOINC,若你也想加入这个计算可以具体查看这一篇教程。 想与各位探讨的是个人觉得若单独拿一段序列进行从头计算然后拼接进模型中是不准确的,更好的方法是进行loop环的补全(若缺失部分为loop环),或者进行分子动力学模拟来预测。 官方的从头预测方法是采用的T4 溶菌酶(噬菌体中)的序列来进行的蛋白结构预测。并于解析了结构的蛋白PDB文件进行比较。
图文无关,纯粹好看
1.准备输入文件
需要:
- fasta格式的序列
- 9mer 片段文件
- 3mer 片段文件
- 已知的pdb文件
1.保存蛋白序列文件为FASTA格式
例如:
|
|
input_files/aa2LZMA03_05.200_v1_3 (3mer fragments) input_files/aa2LZMA09_05.200_v1_3 (9mer fragments)```
这个片段文件生成较为复杂,需要用perl脚本和库文件,安装许多包才能实现,我也还没有完全搞透,由于时间关系,搞透以后作为补充分享给大家。并且搞透的意义不大,毕竟也不是做这个的。 建议大家还是用上面的网站的Fragment Libraries进行在线制作。
3.为了与已知文件进行比较分析结果准确度,所以插入已知文件,当然这个不是必须的。
|
|
由于option文件设置较多,我们分开来讲解。 首先是输入文件设置
|
|
这个没有什么好说的,接下来是建模设置
|
|
increase_cycles表示AbinitioRelax 循环的次数,若想快速完成可以设置为0.1,后面三个参数分别为回转半径,螺旋和环赋予的权重,没有深入了解一般默认即可 然后是relax设置,若想快速完成该步可以不设置
|
|
-fast设置表示在建模后对蛋白进行一个FastRelax,其同时兼容了准确性和速度。 最后是输出
|
|
官方教程设置了两次-nstruct,原因待考。
2.运行Rosetta AbinitioRelax 应用
|
|
当然也可以在后台运行:
|
|
每个结构约运行10-20分钟 没有错误的话将会输出如下结果:
S_00000001.pdb (生成的模型) score.sc (展示得分,Rosetta的得分算法将会在后期介绍) 为了得到精确的结果,建议生成50,000到100,000个模型(官方教程中一说至少1,000)
3.分析结果
3.1 绘制得分图和rmsd图
你可以绘制得分最好的5%或10%的模型图。total_score和rms数据都在score文件中提供。你可以使用同源蛋白做参考(毕竟有pdb数据的话就不用从头建模了)如果连同源结构都没有的话一般考虑最低能量模型。 可以用awk简单的提取数据
|
|
然后可以对数据进行绘图,绘图的方法多种,也非常简单,后期会完整介绍。
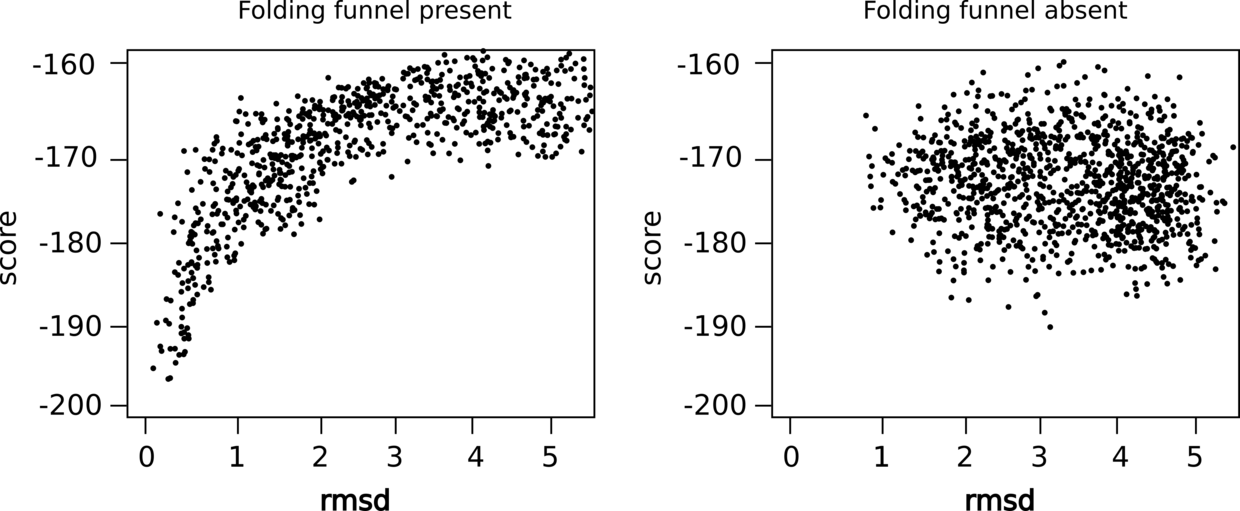
左图可以发现rmsd与score成相关性并且收敛,而右侧没有收敛,说明右侧的结果并不那么可靠
3.2 提取模型
获得的结果中有一个二进制silent file(此要在设置中设置,这样生成的模型都压缩在一个文件中,可以节省空间更加美观,其实也可以不对其进行这个操作,详见Rosetta silent file一章)我们可以从中提取pdb文件,例如我们想提取得分最好的5个文件。
|
|
将会获得模型的名称,将silent file对这些模型名称进行提取即可。
|
|
好的Rosetta得分并不代表结构就一定是好的,所以我们在进行任何分子模拟的时候一定要有许多辅助的实验验证,这样的结果才可是可靠的可信的。我们需要不断的提醒自己。结果可信吗!