转载请联系,且保留链接
PLUMED是许多分子动力学模拟包的插件,主要能进行MD的增强采样或者执行非常多的自由能计算方法。 可以使用诸如元动力学,伞形采样和基于 Jarzynski 方程的转向 MD 之类的现有技术来执行自由能计算作为许多次序参数的函数,特别关注生物学问题。模拟的高分文章很多运用了该工具。
下载地址如下:
plumed-2.4.1
快速安装
如果你迫不及待的想练手,可以通过如下方法简单野蛮式安装:
1
2
3
4
|
./configure --prefix=/usr/local
make -j 4
make doc
make install
|
由于PLUMED需要分析额外的轨迹,同时运行Lennard-Jones 代码,所以需要对模拟的软件进行打补丁,简单的方法如下:
1
2
|
cd /md/root/dir
plumed patch -p
|
当然还是建议按照下面的一步一步的来:
推荐安装
安装BLAS与LAPACK
安装PLUMED依赖的blas和lapack包。(虽然PLUMED自带这两个包,但是网站上说为了性能建议自己编译,未测试自己编译使用和使用自带的性能差异)
1
2
3
|
# 在用户主目录下src下操作
mkdir ~/src
cd ~/src
|
安装BLAS
1
2
3
4
5
6
7
|
wget http://www.netlib.org/blas/blas.tgz
tar zxf blas.tgz
# 可能版本有差别
cd BLAS-3.8.0/
# 编译
gfortran -O3 -std=legacy -m64 -fno-second-underscore -fPIC -c *.f
|
后续工作:
1
2
3
4
5
|
ar r libfblas.a *.o
ranlib libfblas.a
rm -rf *.o # 清理文件
export BLAS=~/src/BLAS-3.8.0/libfblas.a # 导出BLAS环境变量
|
安装LAPACK
1
2
3
4
|
wget http://www.netlib.org/lapack/lapack.tgz
tar zxf lapack.tgz
# 可能版本有差别
cd lapack-3.8.0/
|
编译:
1
|
cp INSTALL/make.inc.gfortran make.inc
|
编辑make.inc文件,大致修改为如下:
1
2
3
4
5
6
7
8
9
10
|
FORTRAN = gfortran
OPTS = -O2 -frecursive -fPIC -m64
DRVOPTS = $(OPTS)
NOOPT = -O0 -frecursive -fPIC -m64
# Define LOADER and LOADOPTS to refer to the loader and desired
# load options for your machine.
#
LOADER = gfortran
LOADOPTS =
|
编译:
后续工作
1
2
|
make clean # 清理文件
export LAPACK=~/src/lapack-3.8.0/ # 导出LAPACK环境变量
|
正式安装
1
2
|
tar xzf plumed-2.4.1.tgz
cd plumed-2.4.1
|
完整的参数可以使用如下命令:
- 官方建议安装在默认预定义位置
1
2
3
4
|
./configure
sudo make -j 4
make check
sudo make install
|
- 当然也可以修改路径:
1
2
3
4
|
./configure --prefix=$HOME/opt
make -j 4 #核数
make check
make install
|
设置环境变量:安装后有设置环境变量提示,里面说直接运行如下tcl脚本即可,但我尝试了未成功
1
2
|
To create a tcl module that sets all the variables above, use this one as a starting point:
@patch/plumed/modulefile
|
我使用的设置如下:
1
2
3
4
5
6
7
8
9
10
|
# plumed
# PLUMED2_HOME内容需要修改成自己的
export PLUMED2_HOME=/to/pacth/plumed
export PATH=$PLUMED2_HOME/bin:$PATH
export LD_LIBRARY_PATH=$PLUMED2_HOME/lib:$LD_LIBRARY_PATH
export PKG_CONFIG_PATH=$PLUMED2_HOME/pkgconfig:$PKG_CONFIG_PATH
export PLUMED_VIMPATH=$PLUMED2_HOME/vim:$PLUMED_VIMPATH
export INCLUDE=$PLUMED2_HOME/include:$INCLUDE
# 若开启了时间计算,需要增加
export PLUMED_USE_LEPTON=yes
|
仅供参考
卸载
在plumed的安装目录执行如下:
更新MD包
现在plumed默认支持如下包(还有一些可以靠插件解决,详情见官网):
- amber14
- gromacs-2016-5
- gromacs-2018-1
- gromacs-4-5-7
- gromacs-5-1-4
- lammps-6Apr13
- namd-2-8
- namd-2-9
- qespresso-5-0-2
- qespresso-6-2
方法很简单,在MD安装的根目录,执行:
!!!!之前安装的话需要重新安装!!!!
在这里简单介绍gromacs的重新安装,具体可以查看之前的文章
1
2
3
4
|
cmake .. -DGMX_BUILD_OWN_FFTW=ON -DGMX_GPU=ON -DGMX_MPI=ON -DCMAKE_INSTALL_PREFIX=/home/yaolab/install/gromacs
make -j 4
make check
make install
|
最后设置环境变量
安装中可能遇到的问题
1
|
WARNING: You might have problems linking FORTRAN programs.
|
但是这个警告是没有关系的,除非你想通过FORTRAN来进行plumed patch --static,那么可以使用参考资料7来进行解决.一般情况下是不需要修改的
简单的总结解决办法就是搜索:libstdc++.so或者libc++.so,在编译的时候设置: LDFLAGS=-L@path其中@path为路径
- 注意:若mpi为自己安装在自己的用户目录的话,那么
sudo mpic++可能会提示找不到,那样的话需要sudoers中增加你的openmpi路径,例如我的:
1
|
/home/kangsgo/install/openmpi/bin/
|
具体可以参考参考资料2-5
- 我在执行
make check时结果如下:
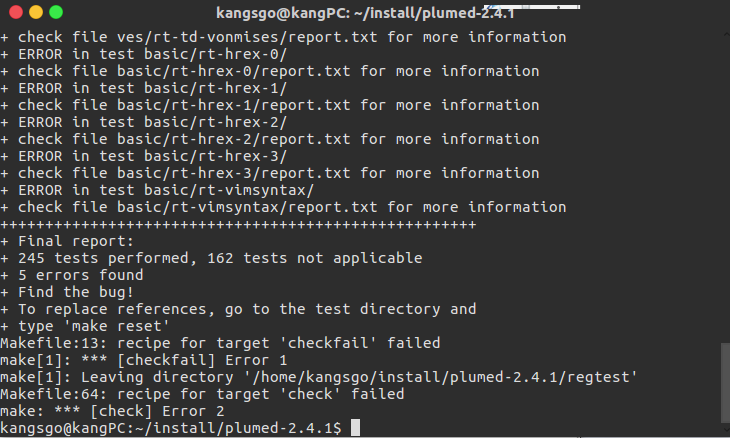
我认为是正常的,因为部分模块默认是没有编译的,具体可以参考参考资料8
参考资料:
- 编译安装BLAS和LAPACK
- mpic++ command not found
- 如何解决sudo命令找不到环境变量的问题
- sudo 环境变量继承和详解
- sudo执行脚本找不到变量
- gromacs + plumed 编译安装教程
- Trouble compiling PLUMED 2.4.0 on Linux (Ubuntu 14.04.5)
- List of modules
- “plumed –has-matheval” with lepton