阅读前请注意:
1.此文仅是我看Martini文献的一些总结,若需要使用Martini力场建议仔细的查看官网信息和相关文献。此文花费较大精力,若有什么意见或者建议欢迎发邮箱交流。邮箱为kangsgo@vip.qq.com 湖大-小康
2.由于我只关注蛋白模拟,故只查阅了Martini力场2.2的文献以及蛋白参数文献
3.此笔记对于Martini性能评测部分只关注了部分内容 Martini力场自问世以来就受到了很多人的喜爱,特别是蛋白的力场,最近(2013年)Martini力场在许多方面都有应用。包括如下:1.蛋白调节囊泡的融合.2.膜结构域的形成.3.磷脂的翻转(flip-flopping).4.磷脂和表面行为(?lipid and surfactant phase behavior)5.单磷脂层的折叠.6.膜束缚(?tether)的形成.7.肽和蛋白介导的膜的重构.7.膜蛋白的自组装和磷脂的分类.8.膜蛋白的门控和构象的变化.9.固体上蛋白的吸附作用.10.蛋白纤维的机制研究.10.蛋白的聚集.11.肽的两亲性12.淀粉纤维研究.13.纳米孔吸附和表面湿度.14.纳米孔的跨膜转运和磷脂吸附.15.膜工程.16.多聚物介导的膜黏附和束缚(?tether).17.nanocoating of a polymer matrix.17.蛋白配体的结合.18.脂蛋白颗粒和纳米圆盘的结构和动力学研究.19.药物传递体系等等。
Martini 2.2和Martini 2.0相比较,做了如下内容的提升:
对蛋白力场进行了修复,包括:
(i)新的脯氨酸,苯丙氨酸和色氨酸侧链拓扑,以改善分区自由能(?partitioning free energies)
(ii)引入偏心电荷模型,以更实际地描述相反电荷的残基之间的联系。
以上两条简而言之就是对之前的电荷模型进行很大的优化
(iii)对于极性侧链参数化极性珠子以提升在非极性环境中的二聚化和表明的结合(iv)调整键的项目来提升α螺旋的长度和增加多聚丙氨酸和谷氨酸的稳定性。
新版本力场命名为Martini 2.2(若为极性水模型,则为2.2P)
Martini 2.2的评价使用了如下的参考评价内容:
伞状采样(PS:文献中纳入为分区自由能中):
对氨基酸残基侧链类似物(SCAs)进行了跨膜(DOPC膜)伞状采样,方法如同Martini 2.0文献中
分区自由能(Partitioning Free Energy):
SCAs在水和油(癸烷,decane)中的分区自由能使用热力学整合的方法计算。具体的为自由能和相应误差使用GROMACS的g_bar(gmx5以上对应的为 gmx bar)分析工具。分区自由能ΔGpart 为水中的自由能减去油中的自由能。
二聚化自由能(Dimerization Free Energy):
也是进行一个另外一种伞状采样(大致为两个SCAs,拉动其中一个SCAs运动)。后经过如下公式计算ΔGdim:
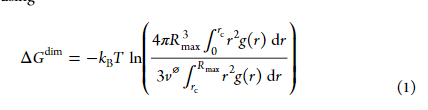
其中kB为波尔兹曼(Boltzmann)约束,T为模拟温度,r为SCAs’ COM距离,rc 是二聚物-单体之间的cutoff(PMF达到第一个最大值的距离),Rmax是考虑的最大距离, v⌀为标准体积(1.66nm3,等于1mol L-1),g(r)为径向分布函数(?回转半径?),其通过PMF可以如下计算:

WW肽结合力(Binding of WW Peptides):
WW表示的为Wimlery-White,是一个五肽,序列为Ac-WLXLL,其中X为可变残基。ΔGWW获得主要是通过free energy perturbation(FEP)和multiple Bennett acceptance ratio(MBAR)相互联系的方法,计算公式为:
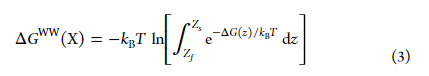
zs=1.0nm,zf=4.0nm,其定义分别为POCC/water限制和bulk water限制。数值使用的Simpson’s 规则计算。相关的X残基ΔΔGWW(X)的计算为ΔGWW(X)减去ΔGWW(Ala)
优化方法和实验结果:
1.改变Phe,Trp和Pro的颗粒类型
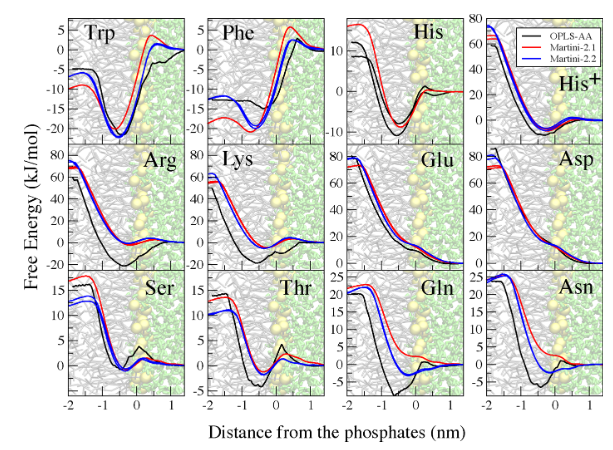
作者第一个方法为通过改变Phe,Trp和Pro的颗粒类型,能够解决Martini 2.1力场中这三个原子类型疏水性太强的弱点,如下表:

其中ref为全原子模拟的结果,通过上表可以发现通过改变颗粒的类型可以使得结果有一定的提升。同时也不会影响伞状采样的结果,对于部分伞状采样的评价相比有更好的结果产生。如下图:
2.改变电荷偏移中心来改善电荷残基
作者通过改变电荷偏移中心来改善电荷残基,在Martini 2.1和Martini 2.1P进行同电荷和异电荷残基的PMF实验时,结果如下图:
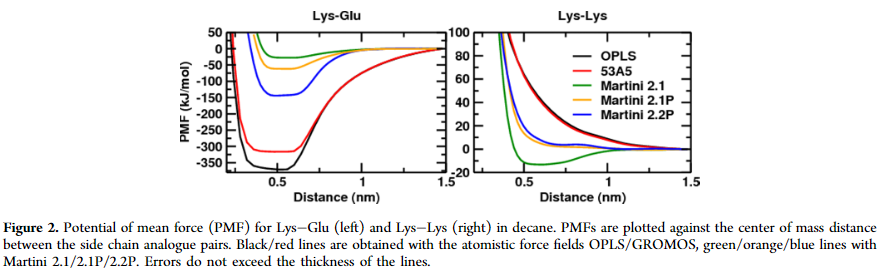
可以发现,在Martini 2.1种,对于带相反电荷的残基仅仅捕捉到微弱的联系,然而,全原子力场表现出非常强的联系。相反,对于相同电荷残基,Martini 2.1预测了稳定的联系,而全原子模型则相反。当使用极性水模型以后(Martini 2.1P),库伦联系下降为1/r,同时因为CG珠子拥有更大的大小(定义的范德华力为~0.26nm)限制了其接近电荷珠子到~0.5nm。然而全原子模型可以更加的接近。
为了弥补这个问题,作者为带电侧链设计了一个替代模型,其中静电和范德华相互作用由两个不同的粒子承载。如下图:
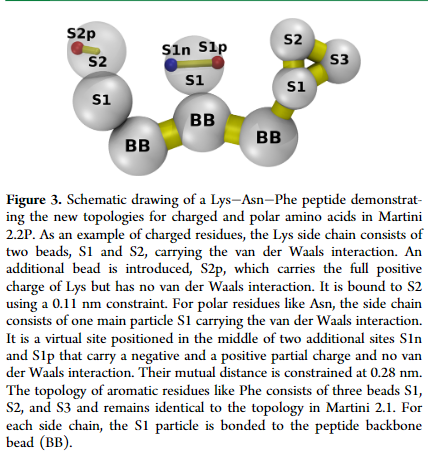
两个颗粒通过一个共约束的0.11nm长的键相互联系。使用这个偏移中心的设置,在Lys-Glu联系对的情况下,电荷可能更接近,并且相互作用增加约3倍(如图2)。但Lys-Lys联系对没有得到改善。
对于其它珠子是否有改善还有待检验。
同时作者还做了以下优化,由于自己用不到,仅在这里做简单介绍:
- 提升非极性残基使用非极性颗粒
- 提升骨架α螺旋和Ala和Gly多聚物序列的稳定性
3. 在溶解肽中测试新的参数
作者进行了一条16肽的模拟(GCN4亮氨酸拉链,PDB code:2ZTA),8号位(Lys)和12号位(Leu)残基突变为非极性的Asn,突变后的肽总共包含5个非极性氨基酸残基(Gln4, Asn8, Asn12, Ser14, Asn16)和6个电荷残基(Lys3, Glu6, Asp7, Glu9, Glu10 and Lys14),统计COM距离结果如下图:
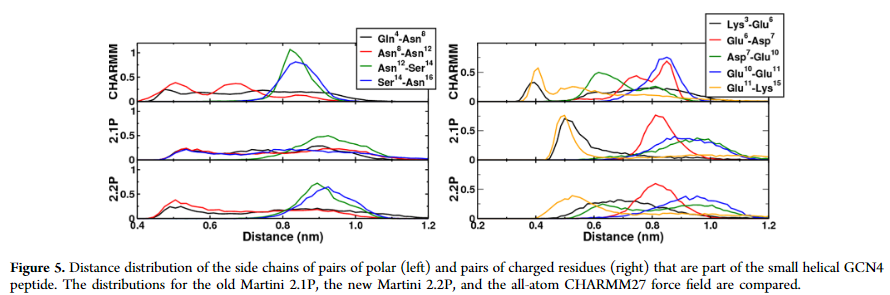
对于非极性氨基酸残基,Martini 2.1P和Martini 2.2P之间有两个方面不同,第一点,峰在短的距离(侧链指向彼此)和长的距离(侧链远离彼此),在Martini 2.2P中转变为一个更小的平均距离,这和全原子模拟更加匹配。第二点为分布更窄(Asn12−Ser14 and Ser14−Asn16)。对于带有相反电荷残基的Lys3-Glu6和Glu11-Lys15联系对,当使用Martini 2.2P时,在短距离处观察到的峰值变得更宽或完全消失。 对于带有相反(Glu6-Asp7)和相等电荷(Glu10-Glu11)direct neighbors,用新参数对相同的方向进行采样。 对于带有相反电荷的Asp7-Glu10残基对,新方向更近的距离被采样到,从而更好地再现了全原子模拟的分布。
虽然这种特定肽的侧链的整体行为似乎已经随着我们的新模型而改善,但是也很清楚,我们的CG模型不能捕获使用原子模型时所看到的分布的一些细节。需要做更多的测试。
文章最后还提到了考虑以后二级结构可以发生转变
参考文献:
- de Jong D H, Singh G, Bennett W F D, et al. Improved parameters for the martini coarse-grained protein force field[J]. Journal of Chemical Theory and Computation, 2012, 9(1): 687-697.