gromacs小分子的配体的电荷问题一直时gromacs模拟蛋白-配体复合物中的难点与重点~网上有许多相关方面的教程,但是都没有成为系统,应李继存老师之邀,把最近摸索的方法过程总结出来,分享给大家~感谢网络上的朋友对我的帮助~
我们以我现在在做的蛋白配体复合物进行模拟,蛋白质为AhR的PAS-B结构域,其是AhR转录因子的配体结合区~TCDD复合物是AhR的经典激动剂。
由于AhR晶体结构并未解析,建模的方法我在这里不累述
你可以在这里下载初始文件:tcddPhe289Ala.tar
mdp文件建议从压缩包中的进行修改,以免直接复制导致换行问题。
1.高斯输入文件建立
1
|
antechamber -i tcdd-h.mol2 -fi mol2 -o tcdd.gjf -fo gcrt -pf y -gm "%mem=1024MB" -gn "%nproc=2" -nc 0 -gk "#HF/6-31G*SCF=tight Test Pop=MK iop(6/33=2) iop(6/42=6) iop(6/50=1) opt nosymm" -ge tcdd_resp.gesp -gv 1
|
其中nosymm是是否保留坐标,可以不加。高斯windows版本内存最多使用1G
2.运行高斯
3.拟合成resp文件
1
|
antechamber -i tcdd_resp.gesp -fi gesp -o tcdd.mol2 -fo mol2 -pf y -c resp -rn TCD
|
4.生成缺失参数文件
1
|
parmchk2 -i tcdd.mol2 -f mol2 -o tcdd.frcmod
|
5.生成AMBER参数文件及坐标文件
1
2
3
4
5
6
7
|
source leaprc.ff99SB
source leaprc.gaff
loadamberparams tcdd.frcmod
lig=loadmol2 tcdd.mol2
check lig
saveamberparm lig tcdd.prmtop tcdd.inpcrd
quit
|
命名为leap.in
6.将AMBER文件转换为GROMACS文件
这里有两种办法amb2gmx.pl和acpype.py,一般使用后者
命令如下:
1
|
python2.7 acpype -p tcdd.prmtop -x tcdd.inpcrd -d
|
将会得到MOL_GMX.gro和MOL_GMX.top两个文件为下面做准备
7.准备蛋白质的拓扑
1
|
gmx pdb2gmx -f mAhR.pdb -o mAhR_processed.gro -water tip3p
|
注:群里说tip3p准确度比spec高
力场选择amber99SB 5
8.准备小分子拓扑结构
将得到的TCD_GMX.top中顶部的[defaults],[ atomtypes ],底部的[ system ],[ molecules ]删除,改名为TCD.itp。记得[ moleculetype ]需要被保留!!
复制mAhR_processed.gro,重命名为complex.gro 将TCD_GMX.gro中内容:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
|
1 TCD CL1 1 1.163 -4.344 0.912
1 TCD CL2 2 1.223 -4.088 0.726
1 TCD CL3 3 2.103 -4.309 1.263
1 TCD CL4 4 2.164 -4.054 1.077
1 TCD O1 5 1.637 -4.310 1.076
1 TCD O2 6 1.690 -4.088 0.913
1 TCD C1 7 1.542 -4.258 0.994
1 TCD C2 8 1.568 -4.148 0.914
1 TCD C3 9 1.759 -4.250 1.075
1 TCD C4 10 1.785 -4.140 0.995
1 TCD C5 11 1.417 -4.317 0.992
1 TCD C6 12 1.469 -4.098 0.832
1 TCD C7 13 1.857 -4.300 1.157
1 TCD C8 14 1.909 -4.081 0.997
1 TCD C9 15 1.318 -4.266 0.911
1 TCD C10 16 1.344 -4.156 0.830
1 TCD C11 17 1.983 -4.242 1.159
1 TCD C12 18 2.009 -4.132 1.079
1 TCD H1 19 1.399 -4.402 1.055
1 TCD H2 20 1.490 -4.013 0.771
1 TCD H3 21 1.836 -4.385 1.218
1 TCD H4 22 1.928 -3.996 0.934
|
添加到complex.gro底部数字行上面将顶部1741加22个原子改为1763,保存。
在topol文件中添加小分子的位置限制:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
|
; Include Position restraint file
#ifdef POSRES
#include "posre.itp"
#endif
; Include ligand topology
#include "TCD.itp"
; Include water topology
#include "amber99sb.ff/tip3p.itp"
[ molecules ]
; Compound #mols
Protein 1
TCD 1
|
[注:增加了itp文件,以及molecules增加了小分子,即:]
1
2
|
; Include ligand topology
#include "TCD.itp"
|
9.定义单元盒子
1
|
gmx editconf -f complex.gro -o newbox.gro -bt cubic -d 1.2
|
1
|
gmx solvate -cp newbox.gro -cs spc216.gro -p topol.top -o solv.gro
|
用vmd观察蛋白体系是否在盒子内部

个人感觉盒子可以调小一些~
10.添加离子
教程使用了能量最小化的文件,我这里稍微改了一下
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
|
; LINES STARTING WITH ';' ARE COMMENTS
title = Minimization ; 标题
define = -DFLEXIBLE ;使用柔性水模型而非刚性模型, 这样最陡下降法可进一步最小化能量
; Parameters describing what to do, when to stop and what to save
; 参数描述如何做,当模拟结束后如何保存
integrator = steep ; Algorithm (steep = steepest descent minimization) 最陡梯度法
emtol = 1000.0 ; Stop minimization when the maximum force < 10.0 kJ/mol 当小于多少能量以后停止最小化
emstep = 0.01 ; Energy step size 能量步大小
nsteps = 50000 ; Maximum number of (minimization) steps to perform 最大最小化步骤
energygrps = system ; Which energy group(s) to write to disk 哪个组进行能量最小化
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
; 参数描述怎样寻找邻近的原子,怎么样计算相互联系
nstlist = 1 ; Frequency to update the neighbor list and long range forces 更新近邻列表的频率. 1表示每步都更新
cutoff-scheme = Verlet ; With cutoff-scheme=Verlet and verlet-buffer-tolerance set, nstlist is actually a minimum value and mdrun might increase it, unless it is set to 1 这样设置更加精确(看英文貌似是的)
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.0 ; Cut-off for making neighbor list (short range forces) 短程力近邻列表的截断值
coulombtype = PME ; Treatment of long range electrostatic interactions 计算长程静电的方法. PME为粒子网格Ewald方法, 还可以使用cut-off
rcoulomb = 1.0 ; long range electrostatic cut-off 长程库仑力截断值
rvdw = 1.0 ; long range Van der Waals cut-off 范德华距离截断值
constraints = none ;无位置限制
pbc = xyz ; Periodic Boundary Conditions 周期性
|
我们这里还需要查看topol文件,看一下我们的体系电荷多少,需要补足电荷,可以看出我们需要补4个CL离子
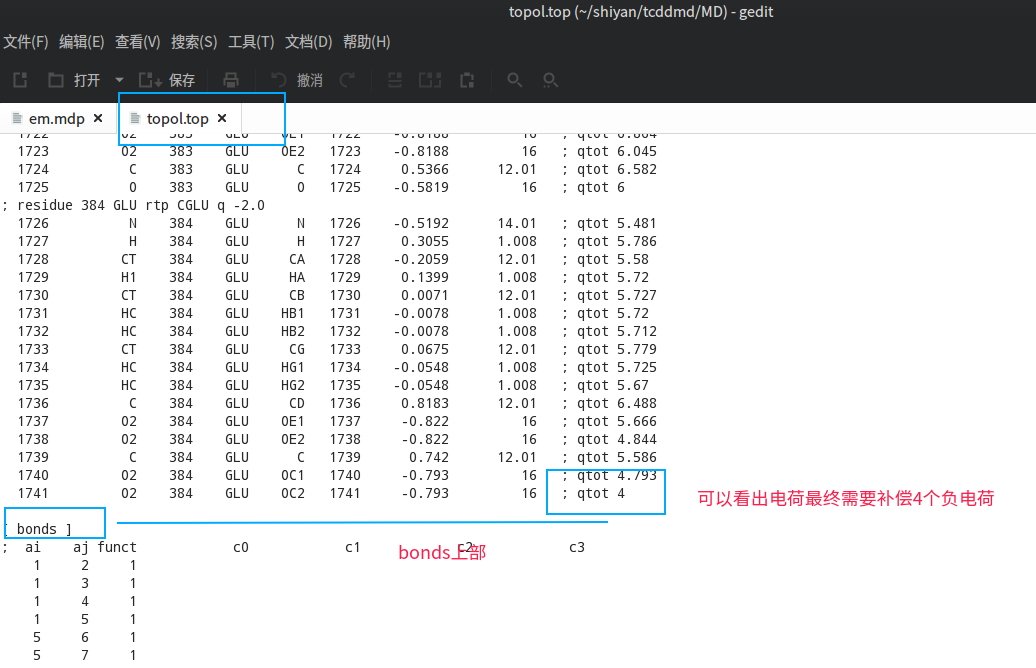
1
|
gmx grompp -f em.mdp -c solv.gro -p topol.top -o ions.tpr
|
做到这里会报错,我找了很久原因,后来查看amber力场文件,发现是大小写不同导致的
我们来看一下我们的TCD.itp文件
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
|
1 Cl 1 TCD CL1 1 -0.072622 35.45000 ; qtot -0.073
2 Cl 1 TCD CL2 2 -0.072622 35.45000 ; qtot -0.145
3 Cl 1 TCD CL3 3 -0.072622 35.45000 ; qtot -0.218
4 Cl 1 TCD CL4 4 -0.072622 35.45000 ; qtot -0.290
5 OS 1 TCD O1 5 -0.320484 16.00000 ; qtot -0.611
6 OS 1 TCD O2 6 -0.320484 16.00000 ; qtot -0.931
7 CA 1 TCD C1 7 0.247670 12.01000 ; qtot -0.684
8 CA 1 TCD C2 8 0.247670 12.01000 ; qtot -0.436
9 CA 1 TCD C3 9 0.247670 12.01000 ; qtot -0.188
10 CA 1 TCD C4 10 0.247670 12.01000 ; qtot 0.059
11 CA 1 TCD C5 11 -0.253268 12.01000 ; qtot -0.194
12 CA 1 TCD C6 12 -0.253268 12.01000 ; qtot -0.447
13 CA 1 TCD C7 13 -0.253268 12.01000 ; qtot -0.701
14 CA 1 TCD C8 14 -0.253268 12.01000 ; qtot -0.954
15 CA 1 TCD C9 15 0.041544 12.01000 ; qtot -0.912
16 CA 1 TCD C10 16 0.041544 12.01000 ; qtot -0.871
17 CA 1 TCD C11 17 0.041544 12.01000 ; qtot -0.829
18 CA 1 TCD C12 18 0.041544 12.01000 ; qtot -0.788
19 HA 1 TCD H1 19 0.196918 1.00800 ; qtot -0.591
20 HA 1 TCD H2 20 0.196918 1.00800 ; qtot -0.394
21 HA 1 TCD H3 21 0.196918 1.00800 ; qtot -0.197
22 HA 1 TCD H4 22 0.196918 1.00800 ; qtot -0.000
|
然而amber99SB力场下如是写的:
1
|
Cl 17 35.45 0.0000 A 4.40104e-01 4.18400e-01
|
以下有两种方法解决:
方法一:
我们需要将top文件的[ atomtypes ]可以写入相应力场的ffnonbonded.itp中,这个方法更加的实用。此方法不建议再使用
个人认为比较稳妥且不会污染力场的方法为将力场文件复制出来,然后在当前目录中进行修改,如果采用apt-get安装,可以在usr/share/gromacs/top下查找对应力场文件,例如拷贝amber99sb-ildn-ligand.ff力场文件,重命名为amber99sb-ildn-ligand.ff,将; Include forcefield parameters下面一行修改为:
1
|
#include "amber99sb-ildn-ligand.ff/forcefield.itp"
|
topol文件其它引用力场的位置可修改可不修改,个人觉得最好修改
方法二:(建议使用该方法)
将[ atomtypes ]写入拓扑中,位于
1
2
|
; Include forcefield parameters
#include "amber99sb-ildn.ff/forcefield.itp"
|
和
1
2
3
|
[ moleculetype ]
; Name nrexcl
Protein 3
|
之间,例如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
|
; Include forcefield parameters
#include "amber99sb-ildn.ff/forcefield.itp"
[ atomtypes ]
;name bond_type mass charge ptype sigma epsilon Amb
c3 c3 0.00000 0.00000 A 3.39967e-01 4.57730e-01 ; 1.91 0.1094
nh nh 0.00000 0.00000 A 3.25000e-01 7.11280e-01 ; 1.82 0.1700
hn hn 0.00000 0.00000 A 1.06908e-01 6.56888e-02 ; 0.60 0.0157
cc cc 0.00000 0.00000 A 3.39967e-01 3.59824e-01 ; 1.91 0.0860
ca ca 0.00000 0.00000 A 3.39967e-01 3.59824e-01 ; 1.91 0.0860
na na 0.00000 0.00000 A 3.25000e-01 7.11280e-01 ; 1.82 0.1700
cd cd 0.00000 0.00000 A 3.39967e-01 3.59824e-01 ; 1.91 0.0860
hc hc 0.00000 0.00000 A 2.64953e-01 6.56888e-02 ; 1.49 0.0157
h1 h1 0.00000 0.00000 A 2.47135e-01 6.56888e-02 ; 1.39 0.0157
ha ha 0.00000 0.00000 A 2.59964e-01 6.27600e-02 ; 1.46 0.0150
h4 h4 0.00000 0.00000 A 2.51055e-01 6.27600e-02 ; 1.41 0.0150
[ moleculetype ]
; Name nrexcl
Protein 3
|
然后就是添加离子了
现在有一种快速添加的方法,即平衡为中性,如下:
1
|
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -neutral
|
当然还有手动平衡的方法如下:
1
|
gmx genion -s ions.tpr -o solv_ions.gro -p topol.top -pname NA -nname CL -nn 4
|
-nn 表示的添加阴离子数目
选择15,添加在水溶剂里面
会有如下话:
1
2
3
4
5
|
Back Off! I just backed up topol.top to ./#topol.top.2#
Replacing solvent molecule 1173 (atom 5282) with CL
Replacing solvent molecule 3252 (atom 11519) with CL
Replacing solvent molecule 2563 (atom 9452) with CL
Replacing solvent molecule 10648 (atom 33707) with CL
|
表示插入进去了
11.能量最小化
参数文件如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
|
; LINES STARTING WITH ';' ARE COMMENTS
title = Minimization ; Title of run
define = -DFLEXIBLE
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 500.0 ; Stop minimization when the maximum force < 5.0 kJ/mol
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
energygrps = Protein TCD ; Which energy group(s) to write to disk
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.0 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; long range electrostatic cut-off
rvdw = 1.0 ; long range Van der Waals cut-off
constraints = none
pbc = xyz ; Periodic Boundary Conditions
|
**能量组为energygrps = Protein TCD,这里TCD需要根据自己的体系进行修改,下同 **
1
|
energygrps = Protein TCD
|
1
|
gmx grompp -f em_real.mdp -c solv_ions.gro -p topol.top -o em.tpr
|
运行模拟
1
|
gmx mdrun -v -deffnm em
|
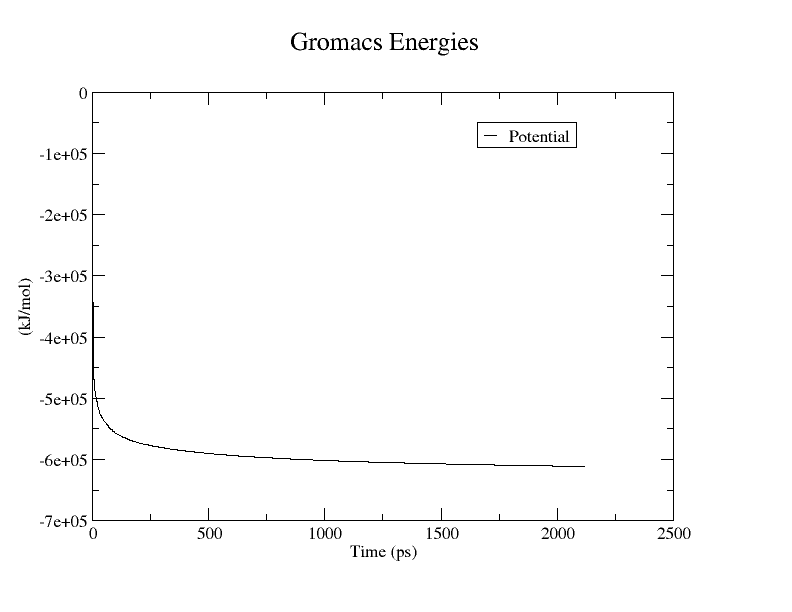
个人认为在运行模拟之前要搞懂参数的意义,所幸这里有中文参考教程,因为要去吃饭了~这里就使用默认的先
我们发现700步就收敛了,所以我决定改成emtol= 1000修改成500再跑````
1888步收敛了~所以我尝试删掉emtol= 1000
但是通过前面的教程,其也是500来步就已经收敛了~
结果如图
PS:作图方法如下:
1
|
gmx energy -f em.edr -o potential.xvg
|
选择 10 0
再用共轭梯度法,mdp文件参数如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
|
; LINES STARTING WITH ';' ARE COMMENTS
title = Minimization ; Title of run
define = -DFLEXIBLE
; Parameters describing what to do, when to stop and what to save
integrator = cg ; Algorithm (steep = steepest descent minimization)
emtol = 100.0 ; Stop minimization when the maximum force < 10.0 kJ/mol
emstep = 0.01 ; Energy step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
nstcgsteep = 1000
energygrps = Protein TCD ; Which energy group(s) to write to disk
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet
ns_type = grid ; Method to determine neighbor list (simple, grid)
rlist = 1.0 ; Cut-off for making neighbor list (short range forces)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; long range electrostatic cut-off
rvdw = 1.0 ; long range Van der Waals cut-off
constraints = none
pbc = xyz ; Periodic Boundary Conditions
|
1
|
gmx grompp -f em2.mdp -c em.gro -p topol.top -o em2.tpr
|
运行模拟
1
|
gmx mdrun -v -deffnm em2
|
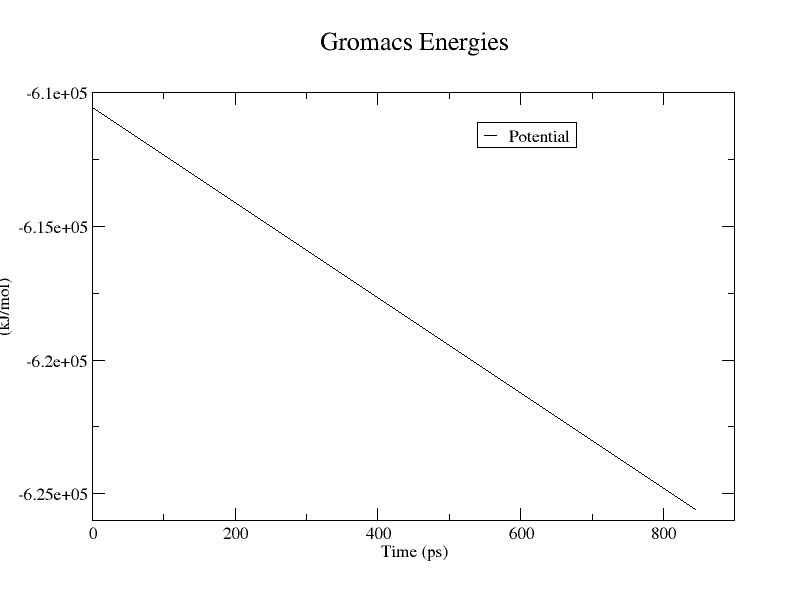
做出来如这个图,图是否正确待考
12.NVT平衡(温度平衡)
限制配体:
1
|
gmx genrestr -f TCD.gro -o posre_TCD.itp -fc 1000 1000 1000
|
在选择组过程中个人觉得选择system或者TCD均可
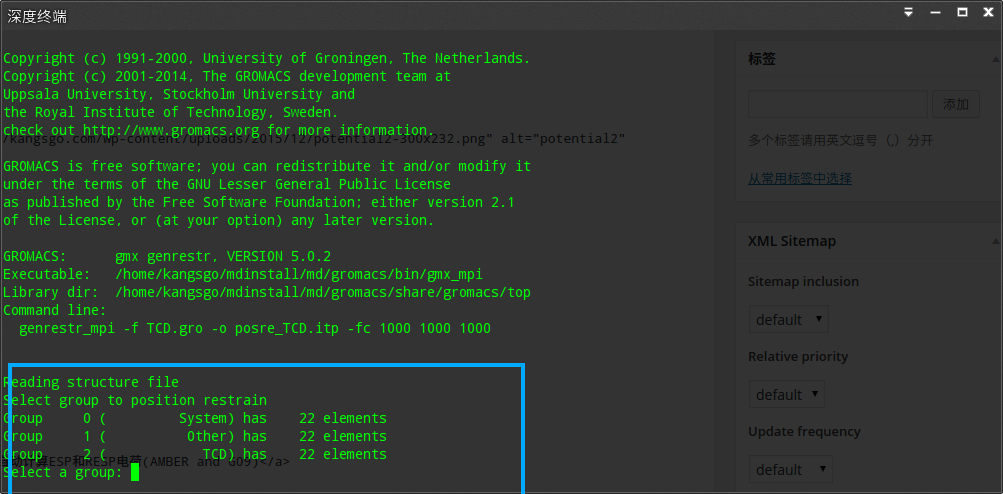
增加配体限制:
1
2
3
4
5
6
7
8
9
10
11
|
; Include ligand topology
#include "TCD.itp"
; Ligand position restraints
#ifdef POSRES
#include "posre_TCD.itp"
#endif
; Include water topology
#include "amber99sb.ff/tip3p.itp"
|
[注:插入的为]
1
2
3
4
|
; Ligand position restraints
#ifdef POSRES
#include "posre_TCD.itp"
#endif
|
其实这里还可以分开限制(具体这里还是没有特别理解~待我看一下参考资料)
热浴
–温度耦合需要蛋白质和配体在一起?
1
|
gmx make_ndx -f em2.gro -o index.ndx
|
选择蛋白和配体分为一组
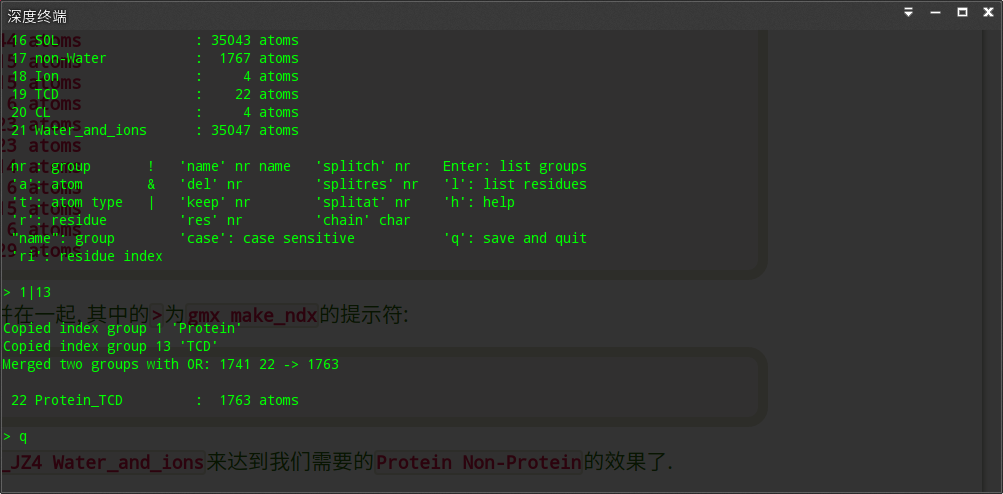
然后就可以热浴了~热浴的mdp如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
|
title = Protein-ligand complex NVT equilibration
define = -DPOSRES ; position restrain the protein and ligand
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 200 ; save coordinates every 1.0 ps
nstvout = 200 ; save velocities every 1.0 ps
nstenergy = 200 ; save energies every 1.0 ps
nstlog = 200 ; update log file every 1.0 ps
energygrps = Protein TCD
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1.4 ; short-range electrostatic cutoff (in nm)
rvdw = 1.4 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein_TCD Water_and_ions ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 300 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed
|
1
2
|
gmx grompp -f nvt.mdp -c em2.gro -p topol.top -n index.ndx -o nvt.tpr
gmx mdrun -deffnm nvt
|
完成后我的析出如图:
1
2
3
4
5
6
7
|
NOTE: The GPU has >20% more load than the CPU. This imbalance causes
performance loss, consider using a shorter cut-off and a finer PME grid.
Core t (s) Wall t (s) (%)
Time: 3799.500 518.255 733.1
(ns/day) (hour/ns)
Performance: 16.672 1.440
|
可以看出我的GPU太渣,暂时没有想到解决办法
我们来看一下NVT耦合的怎么样
1
|
gmx energy -f nvt.edr -o template.xvg
|
选择15 0
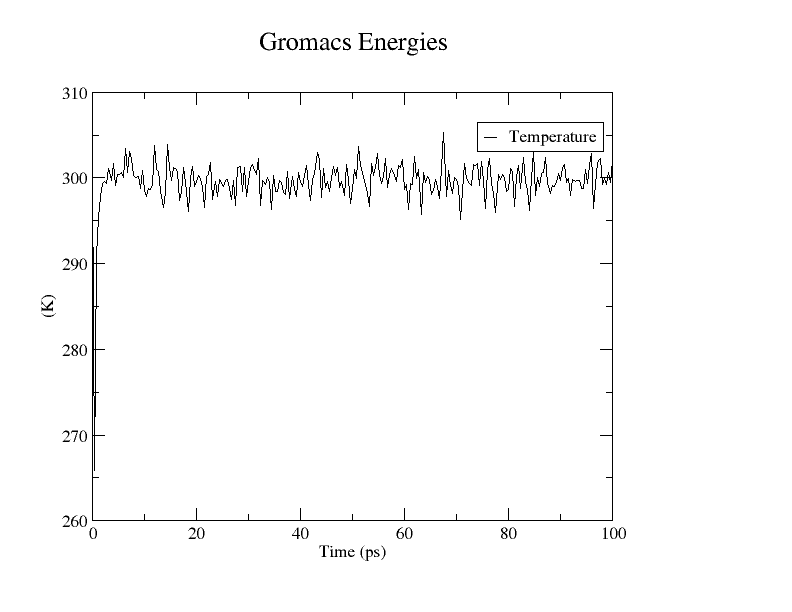
13.NPT平衡
mdp文件如下:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
|
title = Protein-ligand complex NPT equilibration
define = -DPOSRES ; position restrain the protein and ligand
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 200 ; save coordinates every 1.0 ps
nstvout = 200 ; save velocities every 1.0 ps
nstenergy = 200 ; save energies every 1.0 ps
nstlog = 200 ; update log file every 1.0 ps
energygrps = Protein TCD
; Bond parameters
continuation = yes ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1.4 ; short-range electrostatic cutoff (in nm)
rvdw = 1.4 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein_TCD Water_and_ions ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling
pcoupl = Parrinello-Rahman ; pressure coupling is on for NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = no ; velocity generation off after NVT
|
注意,耦合组为Protein_TCD,自己的体系需要修改成自己的,下同
1
2
3
|
gmx grompp -f npt.mdp -c nvt.gro -t nvt.cpt -p topol.top -n index.ndx -o npt.tpr
gmx mdrun -deffnm npt
|
先看一下压力
1
|
gmx energy -f npt.edr -o pressure.xvg
|
选择 16 0
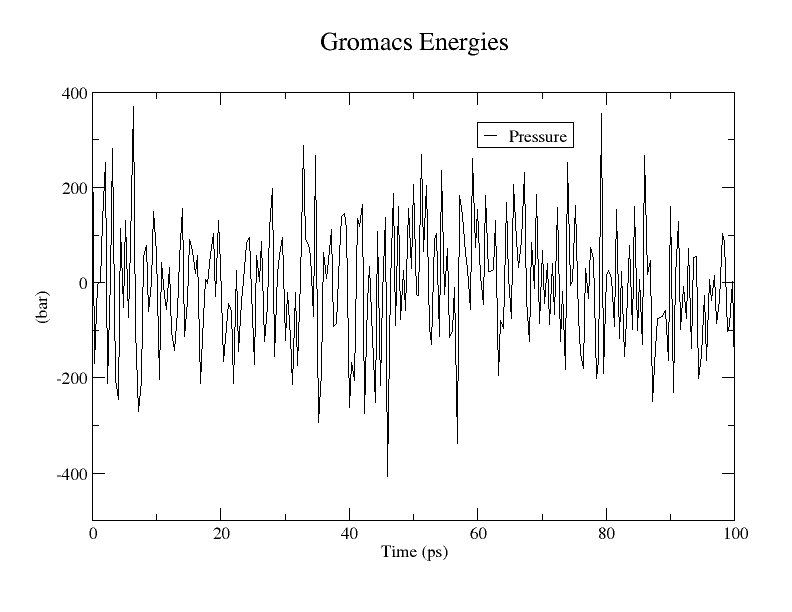
再来看一下密度
1
|
gmx energy -f npt.edr -o density.xvg
|
选择 22 0
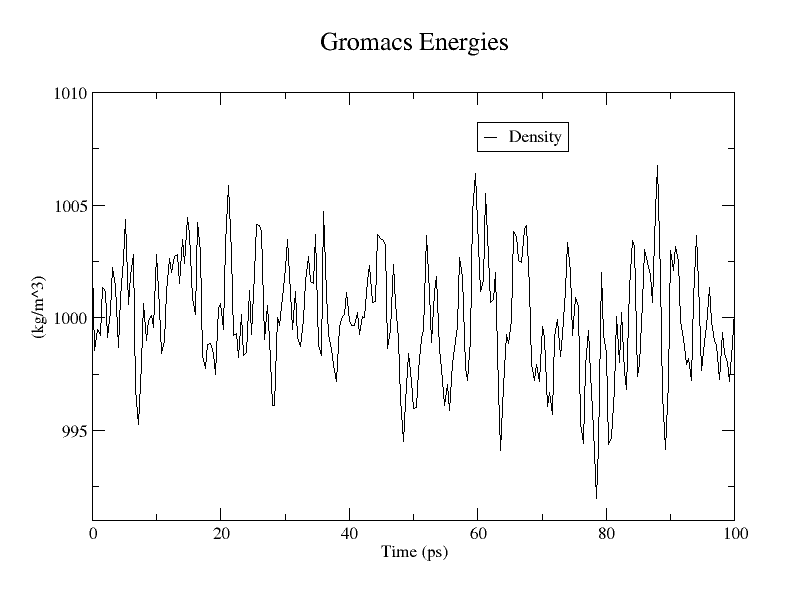
可以看出在1000左右,符合水的密度,个人觉得tip3p效果比spec水模型效果好
14.成品MD
一般MD需要100ns以上,但是我的电脑我怕受不了,就先50ns吧~大约需要3天-4天左右
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
|
title = Protein-ligand complex MD simulation
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 25000000 ; 2 * 25000000 = 50000 ps (50 ns)
dt = 0.002 ; 2 fs
; Output control
nstxout = 0 ; suppress .trr output
nstvout = 0 ; suppress .trr output
nstenergy = 5000 ; save energies every 10.0 ps
nstlog = 5000 ; update log file every 10.0 ps
nstxout-compressed = 5000 ; write .xtc trajectory every 10.0 ps
compressed-x-grps = System
energygrps = Protein TCD
; Bond parameters
continuation = yes ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = all-bonds ; all bonds (even heavy atom-H bonds) constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1.4 ; short-range electrostatic cutoff (in nm)
rvdw = 1.4 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein_TCD Water_and_ions ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling
pcoupl = Parrinello-Rahman ; pressure coupling is on for NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = no ; assign velocities from Maxwell distribution
|
注意:nsteps = 25000000 ; 2 * 25000000 = 50000 ps (50 ns) 这里修改时长,同时注意修改耦合组成为自己的
1
2
|
gmx grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -n index.ndx -o md_0_1.tpr
gmx mdrun -deffnm md_0_1
|
经过2d12h的计算,终于完成了,即高兴又紧张。
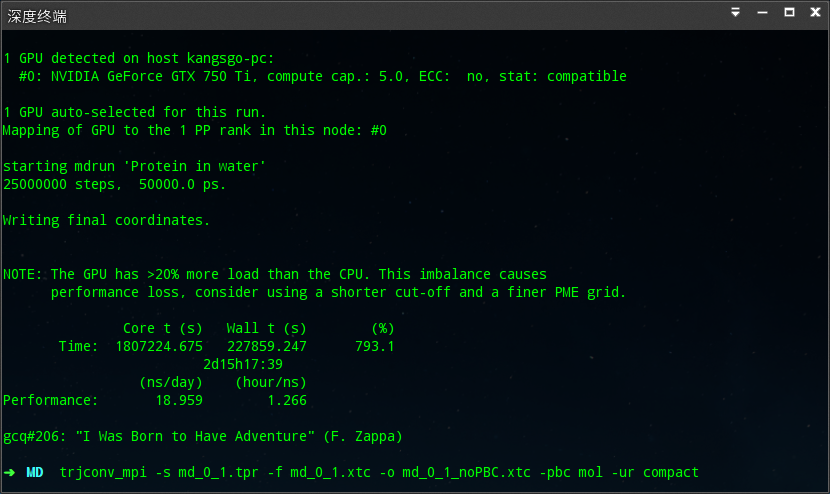
由于这个贴实在太长,另外开一贴
参考文献:自动计算ESP和RESP电荷(AMBER and G09)
参考文献:小分子在GAFF立场GROMCA中的操作
参考文献:使用AmberTools+ACPYPE+Gaussian创建小分子GAFF力场的拓扑文件
参考文献:蛋白质配体复合物