可能很多人喜欢用pymol,其实我认为Chimera是一个更加强大的可视化软件,只是不容易上手,可能不符合国人使用习惯用的人相对较少,这里简单介绍一下最近用到的Chimera补全蛋白缺失结构。
其实Chimera补全蛋白缺失结构主要是是利用的modeller,可以补全尾部结构或者中间的缺失结构,所以个人觉得对于之前介绍的GalaxyFill更加强大。
我们以PDB:1qln为例,1qln为T7 RNA 聚合酶,其中包括了一段核酸序列。我们可以先下载下来了解其信息:
|
|
可以看到MISSING RESIDUES信息,主要是前端和56-71的loop环的缺失。
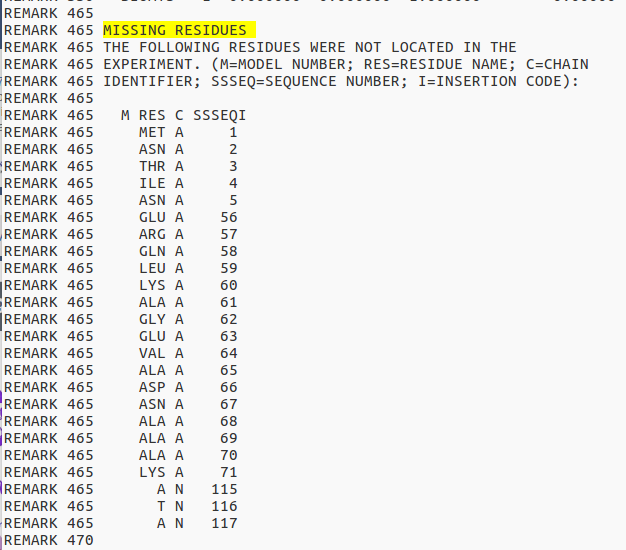
我们将对其中loop进行补齐,若缺失的loop环是自己设计的残基,那么还需要自己修改SEQRES信息,如下图:
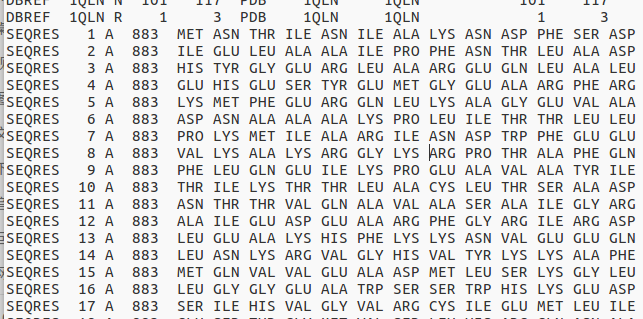 其中
其中SEQRES为完整序列信息(包含缺失序列),可以自己创建或者修改添加从而达到自己的补全内容的目的。除非特殊要求一般PDB数据库中不需要修改或者自己添加。
我们打开UCSF Chimera,点击Favorites -> Command Line,在下面Command中进行下载蛋白,删除核酸等操作:
|
|
 再点击Tools -> Structure Editing -> Model/Refines Loops
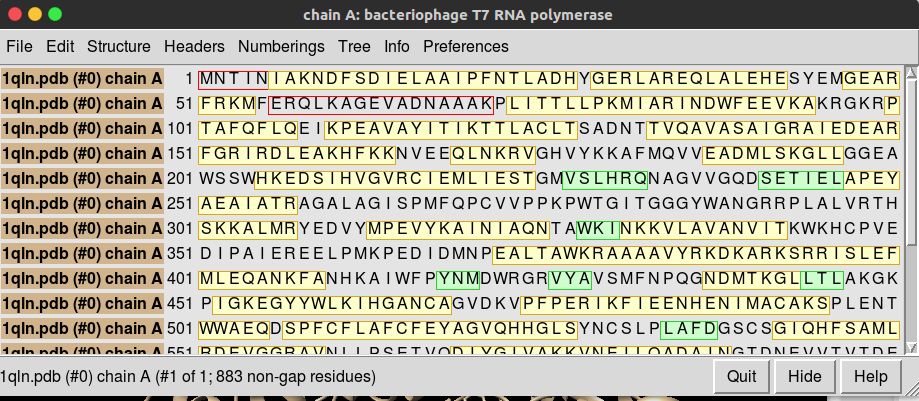
会弹出两个框框,其中这个框框主要是完整的序列信息,其中缺失蛋白位置会用红色框框圈出。
再点击Tools -> Structure Editing -> Model/Refines Loops
会弹出两个框框,其中这个框框主要是完整的序列信息,其中缺失蛋白位置会用红色框框圈出。
 另外一个为设置modeller的框框:
另外一个为设置modeller的框框:
 具体的设置内容如下:
Model/remodel 区域
-active region: 序列框内的活性区域
-Chimera selection region: Chimera中选择的区域
-non-terminal missing structure: 非端点的缺失结构,会将坐标文件和SEQRES相互比较
-all missing structure: 所有缺失片段
**Allow this many residues adjacent to missing regions to move (default 1) **
允许移动的残基(来适配缺失残基),建议就是默认值
Number of models to generate
生成的模型数,个人觉得1个就好,后期再优化,默认为5个
Loop modeling protocol
-standard(默认)
-DOPE: 精度更高,花的时间更多,有可能没有结果或者比预期结果少
-DOPE-HR:和DOPE类似,精度相对没有那么高
Run modeller using
-web service(默认):需要Modeller license key,学术用户可以输入MODELIRANJE
-local installation:需要路径,我的是usr/local/mode9.19
具体的设置内容如下:
Model/remodel 区域
-active region: 序列框内的活性区域
-Chimera selection region: Chimera中选择的区域
-non-terminal missing structure: 非端点的缺失结构,会将坐标文件和SEQRES相互比较
-all missing structure: 所有缺失片段
**Allow this many residues adjacent to missing regions to move (default 1) **
允许移动的残基(来适配缺失残基),建议就是默认值
Number of models to generate
生成的模型数,个人觉得1个就好,后期再优化,默认为5个
Loop modeling protocol
-standard(默认)
-DOPE: 精度更高,花的时间更多,有可能没有结果或者比预期结果少
-DOPE-HR:和DOPE类似,精度相对没有那么高
Run modeller using
-web service(默认):需要Modeller license key,学术用户可以输入MODELIRANJE
-local installation:需要路径,我的是usr/local/mode9.19
点击OK会后台会运行,我运行了大约15分钟,运行完后模型会直接进入界面,然后保存即可。
若停止或者查看可以在Task Panel查看,即右下角 图标
图标